Timm Lankau, Chin-Hui Yu
Computational Evidence for Homonuclear Ge(I)Ge(I) Dative Bonds
J. Phys. Chem. A 124 (2020) 3795 - 3804
→ doi: 10.1021/acs.jpca.9b11665

Dative bonds between identical atoms of the same formal oxidation state are formed as geometric and electronic constraints turn an otherwise lone pair into a bond. Calculations at the B3LYP/6-31+g(d,p) level supported by additional atoms in molecules (AIM) and electron localization function (ELF) analyses on selected germanium polycations are used to prove the concept. The electron density (ρ) is not equally shared between the two atoms of such a homonuclear dative bond (HDB), and the associated charge transfer results in formal charges contradicting chemical convention.
Timm Lankau, Tzu Nung Kuo, Chin-Hui Yu
Computational Study of the Degradation of S-Adenosyl Methionine in Water
J. Phys. Chem. A 121 (2017) 505 - 514
→ doi: 10.1021/acs.jpca.6b09639
The degradation of S-adenosyl methionine (SAM) to homoserine-γ-lactone (HSL) and methyltioadenine (MTA) in water is studied with MD simulations. The AM1 Hamiltonian is used for the quantum part and the flexible AMBER force field for the H2O molecules. The MD simulations predict the free energy barrier for the degradation reaction to be between 109 and 112 kJ mol−1 and an overall gain in free energy of −26 kJ mol−1. The high barrier and the low energy gain of this reaction can be linked to interactions among the carboxylate group of the SAM molecule and solvent H2O molecules, which are not observed on the product side. Hence, the H2O molecules effectively slow down the reaction that otherwise would be much faster.

Timm Lankau, Chin-Hui Yu
Intermediate oxiranes in the base-catalyzed depolymerisation of lignin
Green Chem. 18 (2016) 1590 - 1596
→ doi: 10.1039/C5GC02192H

Monomer yields from the base catalysed depolymerisation (BCD), which has recently evolved as a promising first step in the valorisation of lignin, are severely limited by the repolymerisation of the monomers. DFT calculations with β-phenoxy-α-phenylethanol as a model for the βO4' link in lignin are used to explore various routes for the BCD process and the repolymerisation of its monomers. The depolymerisation proceeds via a resonance-stabilised carbanion, while intermediate oxiranes and formaldehyde, which originates from a second reaction path, are the key compounds in the repolymerisation. It is concluded that controlling the reactive intermediates such as the oxirane in the BCD process will increase monomer yields.
Timm Lankau, Chin-Hui Yu
Solvent effects on the intramolecular conversion of trimethylsulfonium chloride to dimethyl sulfide and methyl chloride
Phys. Chem. Chem. Phys. 16 (2014) 26658 - 26671
→ doi: 10.1039/C4CP03965C
The formation of CH3Cl from (CH3)3SCl in various solvents has been studied with M05/6-311+G(2d,p) DFT calculations to quantify the influence of the solvent on the stability of sulfonium cations. Four different pathways (one SN1, one backside and two frontside attacks for SN2) as well as the formation of different ion pairs (tripod, seesaw, and linear) are discussed to investigate the origin of the kinetic solvent effect (KSE) and the contribution of ion pairs to the overall reaction. Ion pairs are formed only in solvents with a permittivity ε smaller than 28, but the reaction proceeds viaa standard SN2 mechanism with a backside attack in all solvents. The formation of ion pairs does not change the order of the rate law, but it strongly influences the KSE, which can distinguish between reactions starting from the free ions and those starting from ion pairs, in contrast to standard kinetic analysis.

Sheng-Che Yang, Timm Lankau, Chin-Hui Yu
A theoretical study of the nornicotine-catalyzed Mannich reaction in wet solvents and water
Green Chem. 16 (2014) 3999 - 4008
→ doi: 10.1039/C4GC01021C
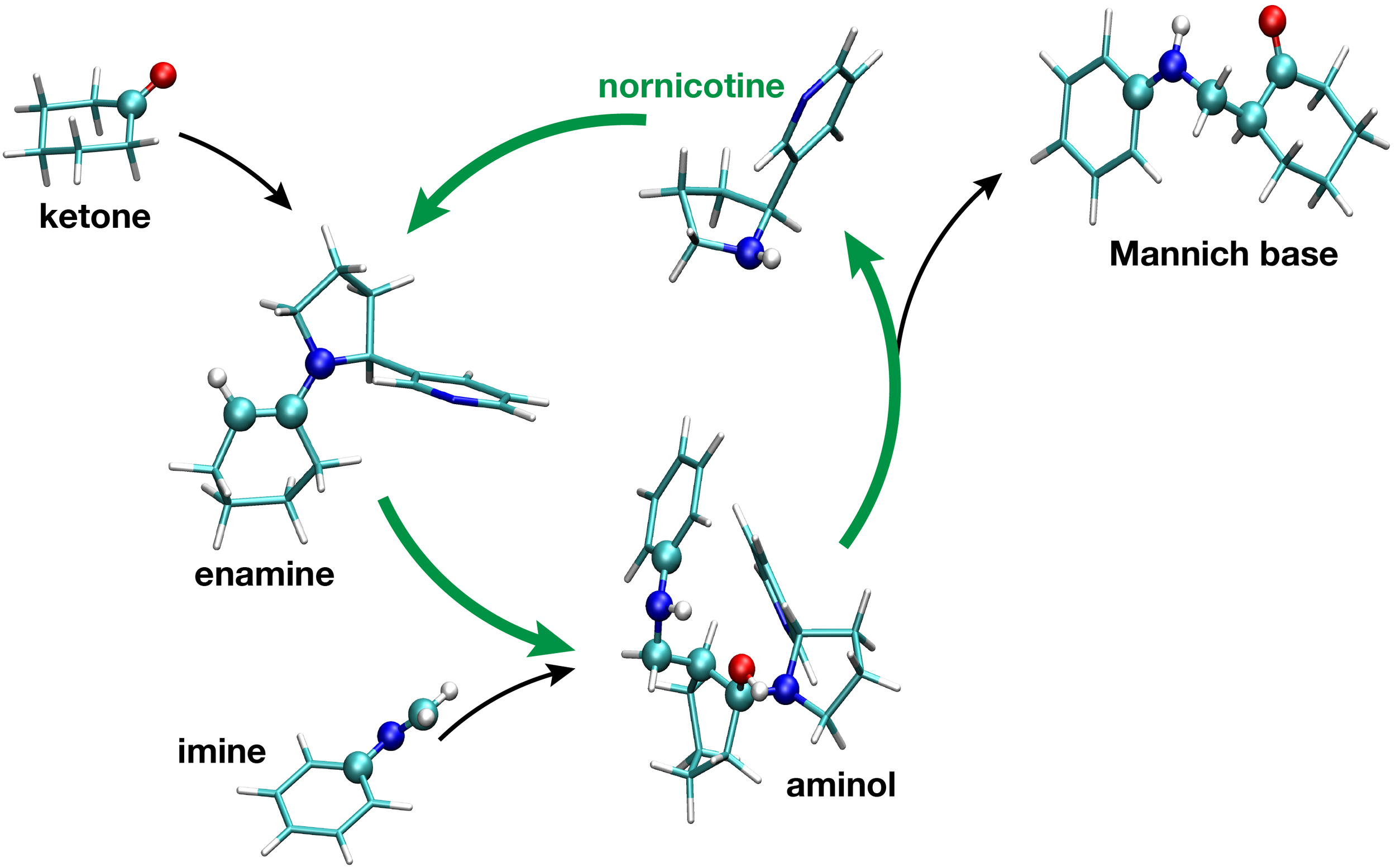
Nornicotine can catalyze the intermolecular Mannich reaction in wet solvents and water to enable a green version of this versatile C-C bond forming reaction. M06-2X/6-31++G(d,p) calculations combined with a polarizable continuum model are used to explore the possibilities of nornicotine catalysis. The bottleneck of the uncatalysed Mannich reaction is the formation of the enol from the ketone, while the catalysed reaction is hindered by the formation of the enamine from the ketone and nornicotine. The calculated barriers for the enamine formation are significantly lower than those for the enol formation and an overall speed-up of the Mannich reaction predicted. The detailed analysis of four possible reaction paths suggests that nornicotine has a mild stereoinductive effect onto the Mannich reaction favoring the S-Mannich product. The analysis further shows that water molecules play an active role in the reaction and indicates that solvents actually need to be wet for nornicotine catalysis.
Timm Lankau, Chin-Hui Yu
A constrained reduced-dimensionality search algorithm to follow chemical reactions on potential energy surfaces
J. Chem. Phys. 138 (2013) 214102 - 214112
→ doi: 10.1063/1.4807743
A constrained reduced-dimensionality algorithm can be used to efficiently locate transition states and products in reactions involving conformational changes. The search path (SP) is constructed stepwise from linear combinations of a small set of manually chosen internal coordinates, namely the predictors. The majority of the internal coordinates, the correctors, are optimized at every step of the SP to minimize the total energy of the system so that the path becomes a minimum energy path connecting products and transition states with the reactants. Problems arise when the set of predictors needs to include weak coordinates, for example dihedral angles, as well as strong ones such as bond distances. Two principal constraining methods for the weak coordinates are proposed to mend this situation: static and dynamic constraints. Dynamic constraints are automatically activated and revoked depending on the state of the weak coordinates among the predictors, while static ones require preset control factors and act permanently. All these methods enable the successful application (4 reactions are presented involving cyclohexane, alanine dipeptide, trimethylsulfonium chloride and azafulvene) of the reduced dimensionality method to reactions where the reaction path covers large conformational changes in addition to the formation/breaking of chemical bonds. Dynamic constraints are found to be the most efficient method as they require neither additional information about the geometry of the transition state nor fine tuning of control parameters.

Timm Lankau, Chin-Hui Yu
A quantum description of the proton movement in an idealized NHN+ bridge
Phys. Chem. Chem. Phys. 13 (2011) 12758 - 12769
→ doi: 10.1039/c0cp02172e

A series of model calculations was done to analyze the delocaliztion of the proton in the linking hydrogen bond of the (Dih)2H+ cation (Dih: 4,5-dihydro-H-imidazole). Standard quantum chemical calculations (B3LYP/D95+(d,p)) predict a low barrier hydrogen bond (LBHB) and thereby a delocalized proton in the NHN+ hydrogen bridge. Explicit quantum calculations on the proton indicate that the delocalization of the proton does not provide enough energy to stabilize a permanent LBHB. Additional Born-Oppenheimer Molecular Dynamics (BOMD) simulations indicate further that the proton is localized at either sides of the NHN+ bridge and that a central proton position is the result of temporal averaging. The possibility of the proton to tunnel from one side to the other side of the NHN+ bridge increases with the temperature as the trajectory of the (Dih)2H+ cation runs through regions where the thermal excitation of Dih ring vibrations creates equal bonding opportunities for the proton on both sides of the bridge (vibrationally assisted proton tunneling). The quantum calculations for the proton in (Dih)2H+ suggest further a broad peak for the 1←0 transition with a maximum at 938 cm-1 similar to that observed for LBHBs. Moreover, the asymmetry NHN+ bridge in a thermally fluctuating environment is strong enough to create a significant peak at 1828 cm-1 for the 2←0 transition, while contributions from the 2←1 are expected to be weak for the same reason.
T. Lankau, C.H. Yu
A Model Study of the Efficiency of the Asp-His-Ser Triad
J. Comp. Chem. 31 (2010) 1853 - 1859
→ doi: 10.1002/jcc.21470
The observation of the Asp-His-Ser triad (Asp: aspertate, His: histidine, Ser: serine) triad both in mammalian and bacterial proteases suggests a special efficiency. A series of B3LYP/D95*(d,p) calculations on various [X-HβY]-} dyads (as part of the [X-HβY-HαAc]- model triad, HAc: acetic acid) made from 8 different anions X- and 15 different coupling elements HβY was done to analyze the molecular origin of this efficiency.
The X- anion acts merely as an electron density donor independent of its chemical nature and the evolutionary selection of Asp for the catalytic triad seems therefore to be caused by the pH of the triads environment. As the linking proton Hβ moves from Y- to X-, electron density is effectively moved from X- to Y- increasing thereby the proton affinity (PA) of the [X-HY]- dyad, which finally leads to the deprotonization of the HAc molecule. The degree to which the position of Hα controls the PA is dominatly determined by the coupling element HY. The model calculations indicate that 4-Methyl-1H-imidazole (HMim) is a very efficient coupling element, which suggest the evolutionary convergence to the Asp-His-Ser is not only controlled by the ready availability of the imidazole motive in His but also by its very high efficiency.

T. Lankau, C.H. Yu
Correlated Proton Motion in Hydrogen Bonded Systems: Tuning Proton Affinities
Phys. Chem. Chem. Phys. 9 (2007) 299 - 310
→ doi: 10.1039/b612945e

The theorem of matching proton affinities (PA) has been widely used in the analysis of hydrogen bonds. However, most experimental and theoretical investigations have to cope with the problem that the variation of the PA of one partner in the hydrogen bond severely affects the properties of the interface between both molecules. The B3LYP/d95+(d,p) analysis of two hydrogen bonds coupled by a 5-methyl-1H-imidazole molecule showed that it is possible to change the PA of one partner of the hydrogen bond while maintaining the properties of the interface. This technique allowed us to correlate various properties of the hydrogen bond directly with the difference in the PAs between both partners: It is possible to tune the potential energy surface of the bonding hydrogen atom from that of an ordinary hydrogen bond (localized hydrogen atom) to that of a low barrier hydrogen bond (LBHB, delocalized hydrogen atom) just by varying the proton affinity of one partner. This correlation shows clearly that matching PAs are of lesser importance for the formation of a LBHB than the relative energy difference between the two tautomers of the hydrogen bond.